Peptide Science
·15 min read
T-Cell Rebirth: Can Modulating CD28 Reverse the Aging of the Human Immune System?
The loss of a single molecule, CD28, is the most consistent clock in the aging human immune system. The provocative question isn't whether it falls with age, it does. It's whether that fall is destiny, or a signal we can read, slow, and one day rewrite.
By Tony Medrano, LongevityPlan.AI
Here is an uncomfortable fact for anyone over forty who has just shrugged off a flu shot that "didn't take," a cold that lingered three weeks, or a relative whose cancer appeared out of nowhere. The competence of your immune system is not a vague abstraction. It can be measured. And one of the most reliable readouts is the gradual disappearance of the costimulatory receptor CD28 from the surface of your T cells.
In a newborn, essentially every naïve T cell wears CD28.[2] By the eighth or ninth decade, large armies of T cells — especially the killer CD8+ lineage — have shed it entirely. These CD28-null cells are not science trivia. Their frequency predicts who responds poorly to vaccination, who carries more chronic inflammation, and, in several cohorts, who fractures and declines.[3] Abbe Vallejo, whose Mayo Clinic group mapped the molecular machinery of this loss, called CD28-null T cells "the most consistent biological indicator of aging in the human immune system."[3]
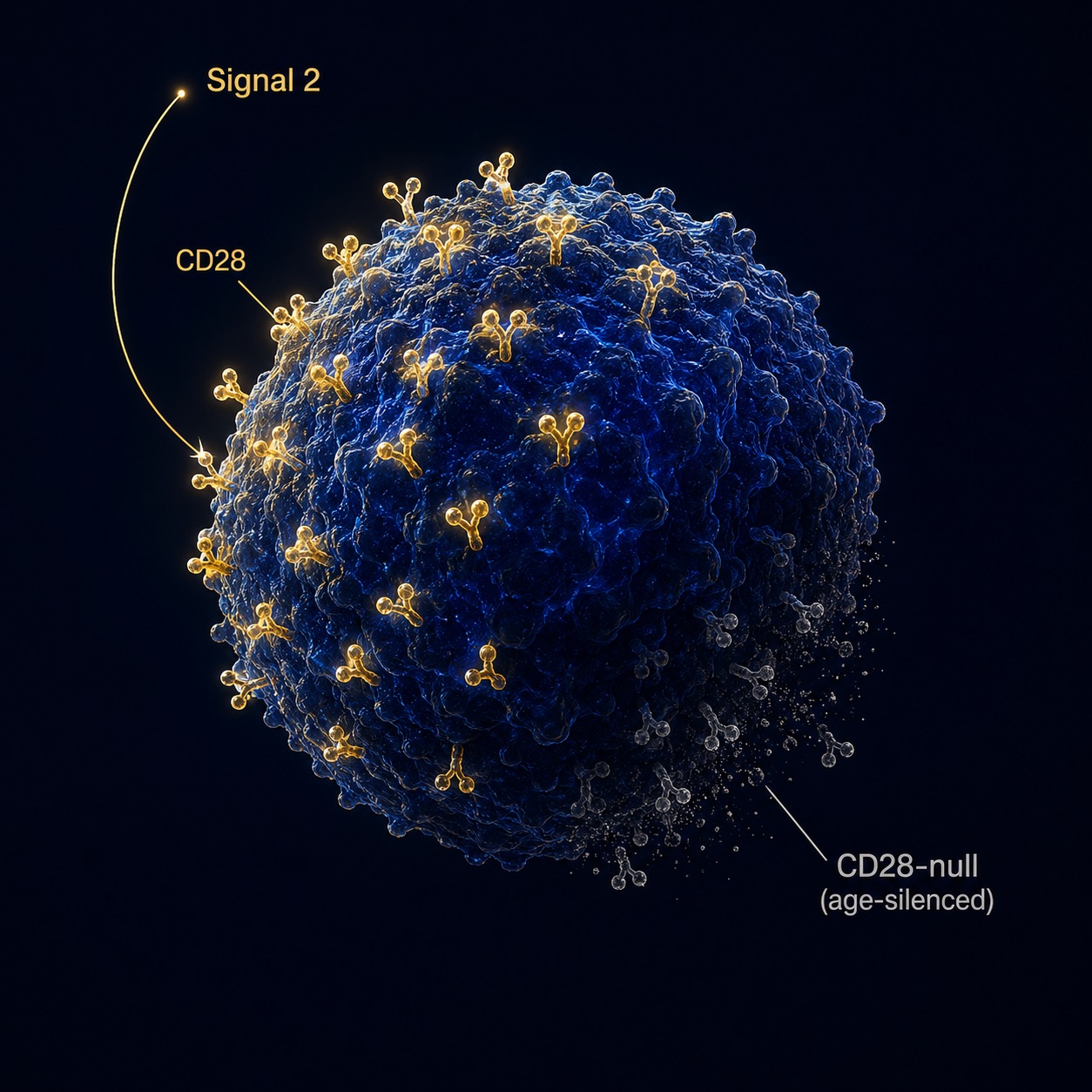
Figure 1. CD28 Costimulatory Receptor on a Human T Cell — The Most Consistent Biomarker of Immune Aging
A healthy T cell wears the CD28 receptor across its surface (gold). With age and chronic infection, it sheds the receptor (grey) — the loss that serves as the single most consistent clock in the aging human immune system.
So the title's question is a serious one, and we will treat it seriously — including the parts where the honest answer is "not yet." This is not a self-help promise that you can will your T cells young. It is a tour of what the data actually show, what the world's cell-therapy laboratories are already doing about it, and what a precision, data-driven longevity plan can responsibly do with that knowledge today.
1. Signal Two: The Molecule That Says "Go"
A T cell deciding whether to attack is a paranoid bureaucrat. It refuses to act on a single piece of evidence. To activate, it demands two signals. Signal one is the T-cell receptor recognizing its specific antigen. Signal two — costimulation — is delivered when CD28 on the T cell binds CD80 or CD86 on an antigen-presenting cell. Without signal two, the cell doesn't just stay quiet; it can be driven into anergy, a state of lasting unresponsiveness.
CD28 is not a mere amplifier. In their definitive 2016 review, Jonathan Esensten and Jeffrey Bluestone of UCSF showed that CD28 ligation triggers epigenetic, transcriptional, and metabolic programs that, in their words, "cannot be recapitulated by TCR ligation alone."[1] Costimulation lowers the activation threshold, fuels the glycolytic burst that powers clonal expansion, and underwrites the survival and differentiation of both effector and regulatory T cells. It is, in short, the permission slip for adaptive immunity — and the longevity story begins when that permission slip begins to disappear.
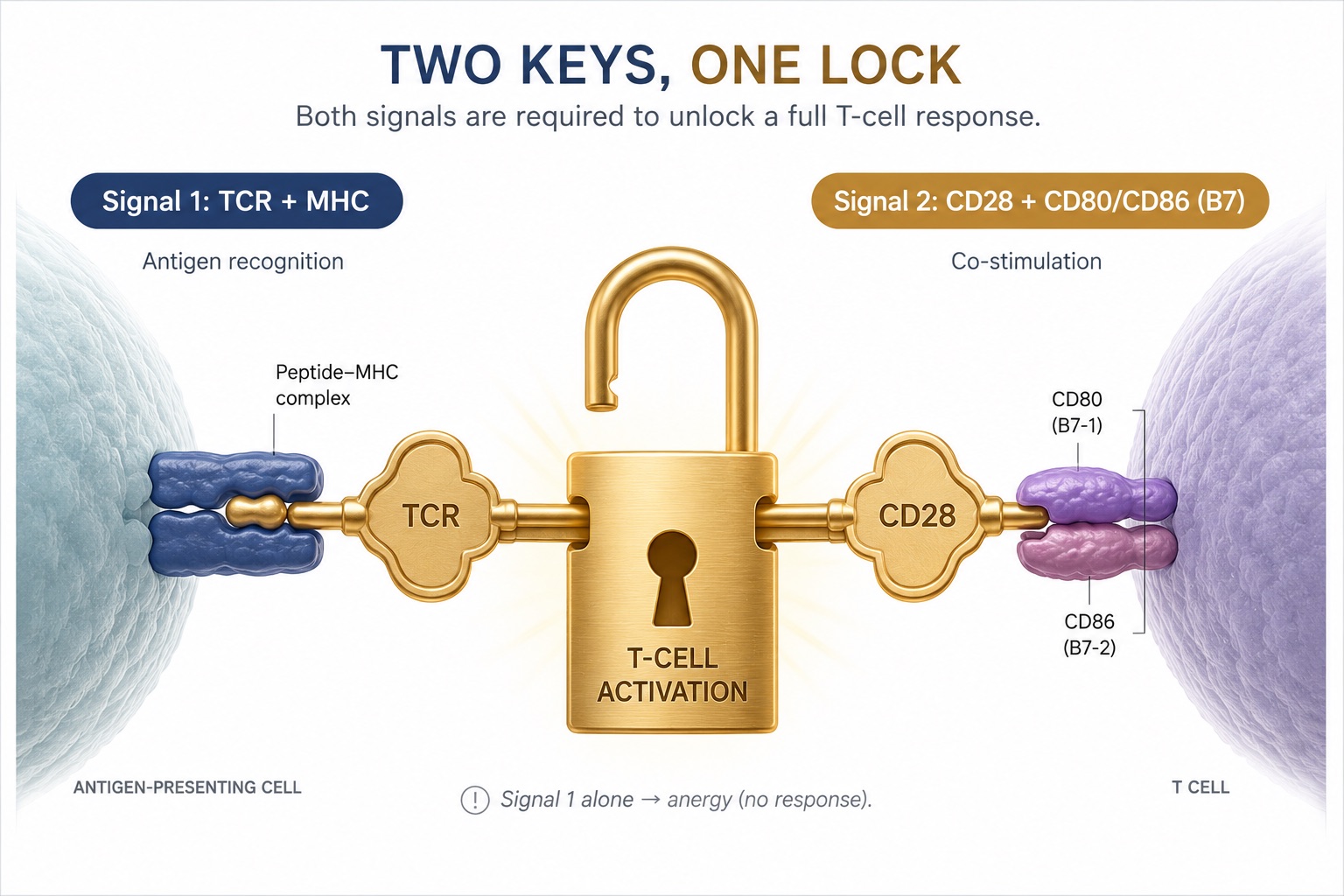
Figure 2. Two-Signal T-Cell Activation — TCR Antigen Recognition Plus CD28–B7 Costimulation
A T cell needs two keys to fire — antigen recognition (Signal 1) and CD28 costimulation (Signal 2). Lose CD28, and even a correctly identified threat can be met with silence.
2. The Most Consistent Clock in the Body
Why does CD28 vanish? The proximate cause is repetition. Every time a T cell is stimulated, CD28 transcription dips and then recovers. But under decades of chronic antigenic pressure — and with one antigen in particular, the near-ubiquitous cytomegalovirus (CMV) — the recovery eventually fails.[2] Vallejo, Weyand, and Goronzy traced the failure to its molecular root: an inoperative transcriptional initiator in the CD28 gene, with inflammatory tumor necrosis factor-α and B7 ligands actively repressing the promoter.[4],[14] The cell doesn't lose CD28 by accident. It is silenced.
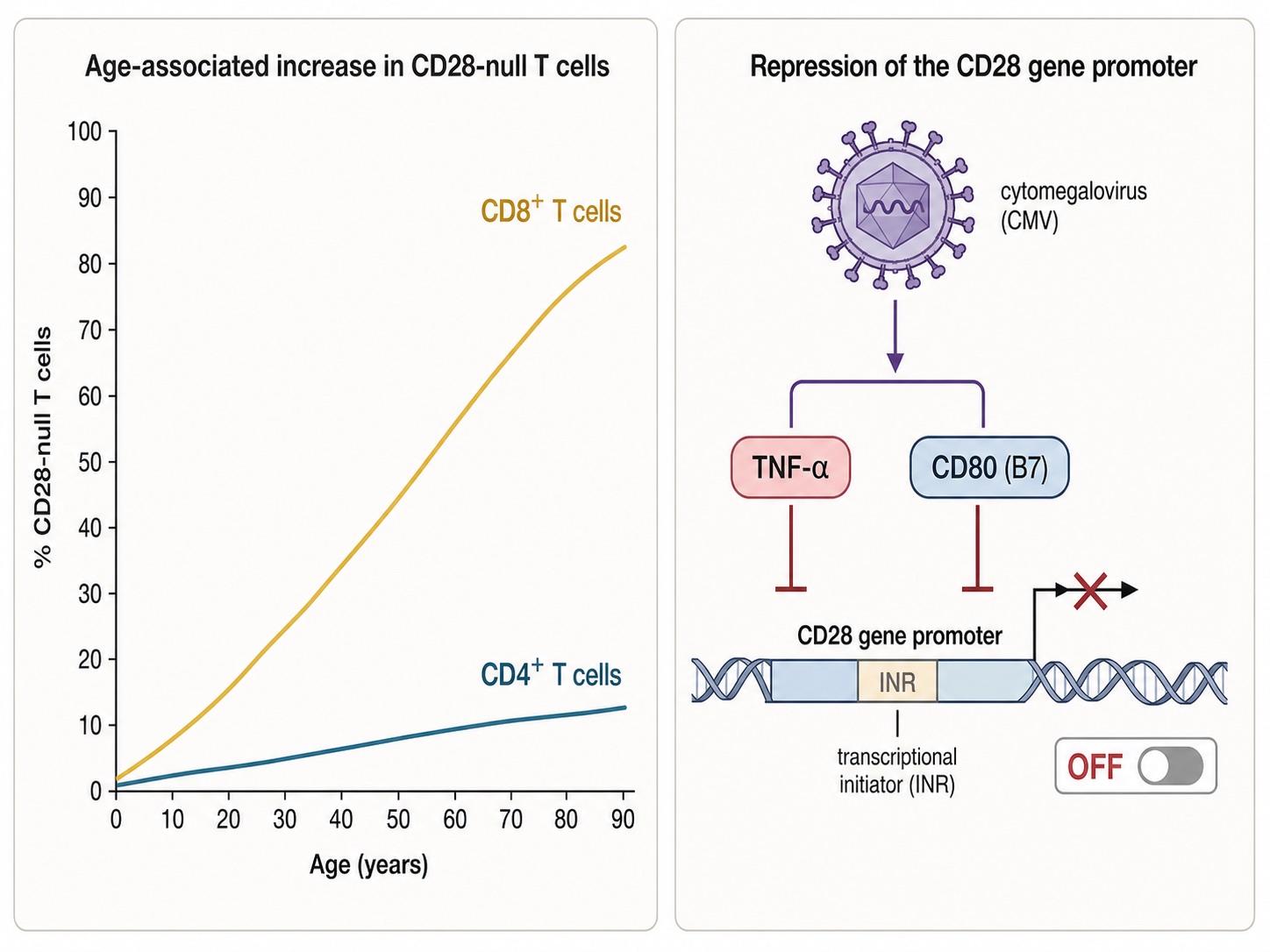
Figure 3. Age-Related Accumulation of CD28-Null T Cells and Transcriptional Silencing of the CD28 Gene
With each decade — and accelerated by lifelong CMV infection — CD8⁺ T cells progressively silence the CD28 gene. Inflammatory signals such as TNF-α actively switch the receptor off; this is regulation, not random decay.
What's left behind is a peculiar cell. Pioneering work by Rita Effros at UCLA established that these CD28-null CD8+ cells bear the fingerprints of replicative senescence: critically short telomeres, resistance to apoptosis, and a loss of proliferative capacity.[6],[7] They are long-lived, oligoclonal, and — crucially — not inert. They are armed cytotoxic cells brimming with perforin and granzyme B, and they pump out inflammatory cytokines. In other words, the aging immune system doesn't simply weaken. It becomes simultaneously less precise and more inflammatory: weaker where you need strength, louder where you need silence.
This is the engine of what gerontologists call inflammaging, and it sits inside the broader framework María Mittelbrunn and Guido Kroemer laid out in their influential 2021 review, "Hallmarks of T cell aging."[9] They cataloged ten molecular hallmarks — from thymic involution to mitochondrial dysfunction — and made a striking argument: T-cell aging is not merely a consequence of getting old. It can be a cause of it. In a landmark 2020 experiment, Desdín-Micó and colleagues engineered T cells with dysfunctional mitochondria and watched them induce multi-organ deterioration and premature senescence across the whole animal.[10] The immune system, it turns out, is not a passenger on the aging journey. Sometimes it is driving the bus.
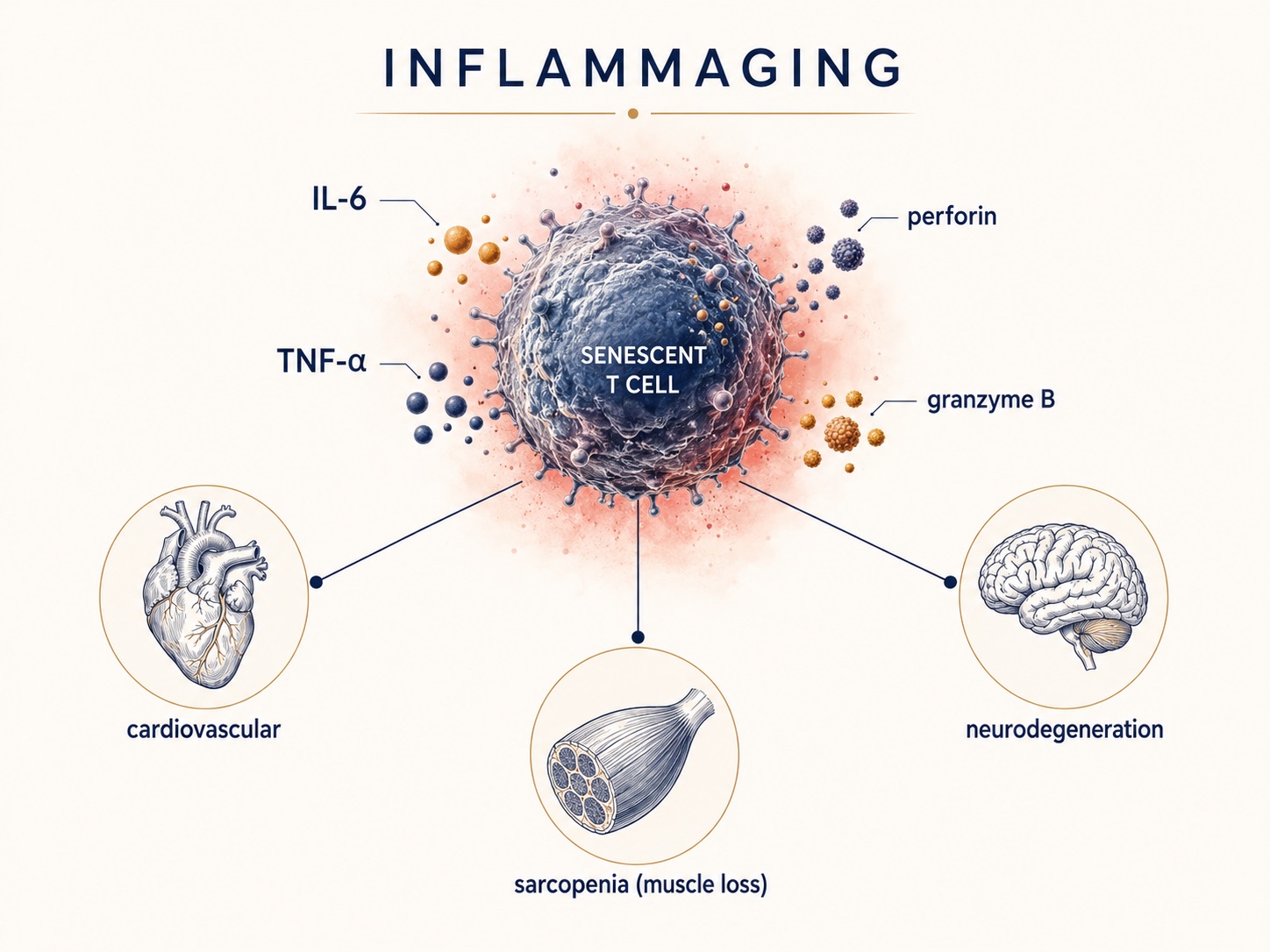
Figure 4. How Senescent T Cells Drive Systemic Inflammation and Age-Related Disease
Aged T cells don't merely go quiet — they smolder, spilling inflammatory signals ("inflammaging") that feed cardiovascular disease, neurodegeneration, and muscle loss. Immune aging sits upstream of the conditions that actually end health spans.
3. Why a Healthy Executive Should Care
Suppose you are a 54-year-old founder who runs half marathons and never gets sick. Three reasons this clock should still interest you.
Cancer surveillance. The same immune cells that should patrol for nascent tumors lose their edge as the naïve repertoire contracts and senescent cells accumulate. Impaired immune surveillance has been shown, in animal models, to accelerate the accumulation of malignant and senescent cells. This is the biological basis for why cancer incidence is, overwhelmingly, a disease of the second half of life.
Vaccine response. A high burden of CD28-null cells correlates with blunted antibody responses to vaccination in older adults.[3],[6] If you intend to rely on a shingles, influenza, or future cancer vaccine when you are seventy, the soil those vaccines land in is being graded now.
Frailty and the inflammatory baseline. Goronzy and Weyand, in their 2019 Nature Reviews Immunology synthesis on T-cell aging mechanisms, link this immune drift to the chronic, low-grade inflammation underlying cardiovascular disease, neurodegeneration, and sarcopenia.[8] The CD28-null clock is not a niche immunology metric. It is upstream of the diseases that actually end healthspans.
4. The Heresy: Is the Clock Reversible?
For two decades, the dogma was that CD28 loss is a one-way street — terminal, irreversible, a tombstone. The interesting science of the last few years is the quiet erosion of that certainty.
The first crack came from the people who discovered the silencing. In 2003, Warrington, Vallejo, Weyand, and Goronzy reported that interleukin-12 could re-induce CD28 expression on senescent CD4+ T cells, restoring elements of helper function.[5] The silenced gene, it turned out, was not deleted. It was switched off — and switches, unlike deletions, can be flipped. Subsequent work showed the loss is governed by reversible regulatory layers: specific DNA-methylation patterns and microRNA programs that distinguish CD28-null cells from their CD28-positive precursors.[15] Epigenetic states are, by definition, modifiable.
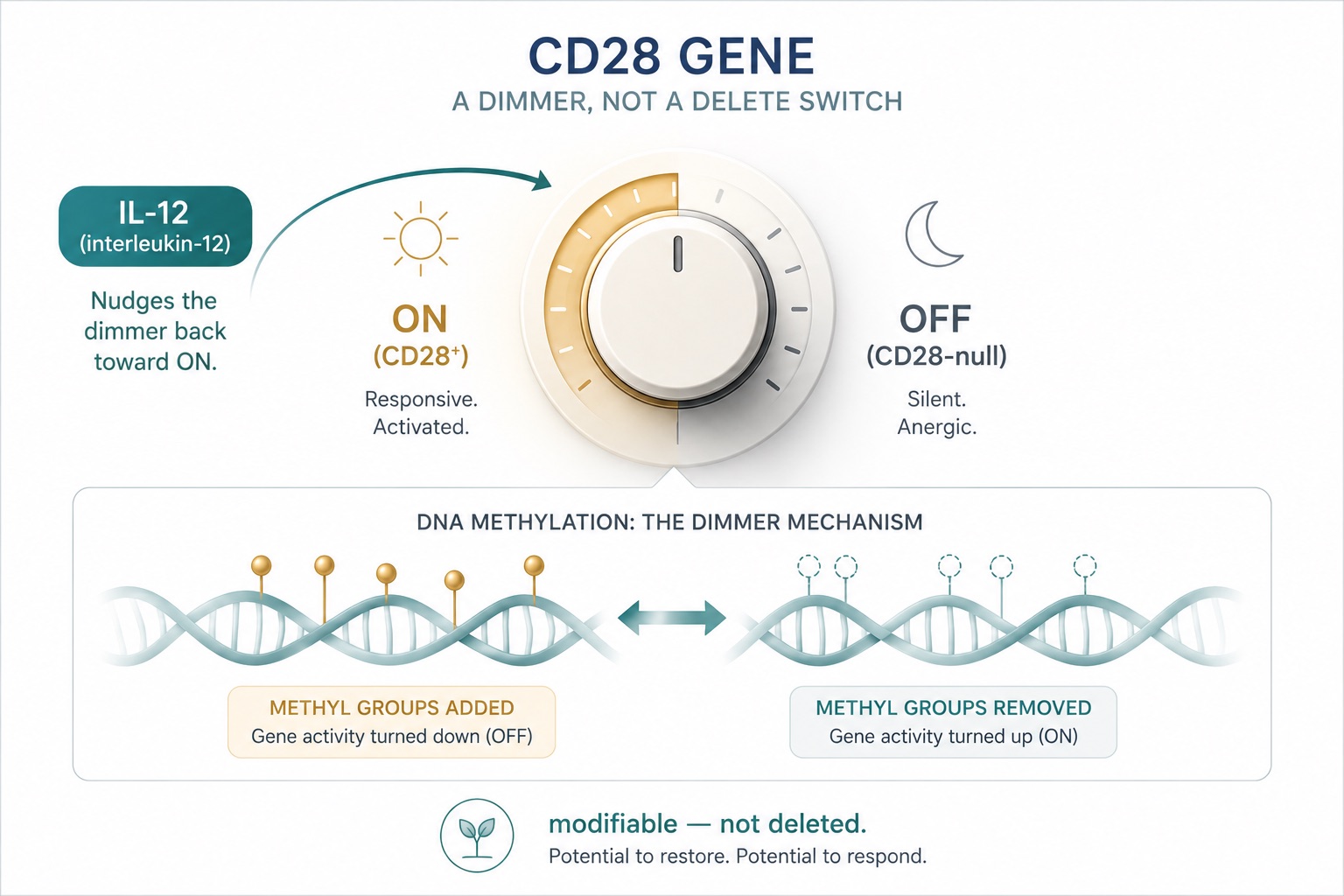
Figure 5. CD28 as a Reversible Epigenetic Switch — Interleukin-12 Re-Induces CD28 on Senescent T Cells
The CD28 gene is switched off, not erased — and switches can be flipped. In the laboratory, interleukin-12 re-induced CD28 on senescent T cells, reframing immune aging as a dial rather than a one-way verdict.
The second crack came from distinguishing two things that had been carelessly merged. Arne Akbar's group at University College London has argued forcefully that senescence and exhaustion are different phenomena with different regulation — and that senescent T cells are not simply dead weight but can be functionally reinvigorated.[11],[12] Akbar's lab showed that blocking the p38 MAPK stress pathway and sestrin signaling modulates the senescent phenotype, in one case switching senescent-like CD8+ cells toward natural-killer-like function.[13] These are not yet therapies. But they convert a fatalistic claim ("immune aging is inevitable") into an empirical research program ("which levers move it, and by how much").
That reframing is the entire thesis of this article. The loss of CD28 is best understood not as an inevitable end-point of cellular aging, but as a modifiable signal — a dial, not a destiny. A 2024 review on CD28-null T cells across aging and disease put the field's two live questions plainly: can this population serve as a clinical biomarker of biological age and adverse outcomes, and can targeting it become an intervention to delay immune aging?[16] Both are now open scientific questions rather than settled negatives. That is real progress.
5. When Nature Deletes CD28, Engineers Put It Back
Here is the part of the story that should make any data-minded person sit up. While aging biology spent decades documenting how the body removes CD28 signaling, an entirely separate field of medicine was quietly building its greatest triumph by reinstalling it.
Chimeric antigen receptor (CAR) T-cell therapy reprograms a patient's own T cells into what Michel Sadelain — who identified CD19 as the foundational CAR target and now directs the Columbia Initiative in Cell Engineering and Therapy — famously calls "living drugs."[20] The first CAR-T therapies were approved in 2017; more than 45,000 patients with otherwise refractory leukemias, lymphomas, and myelomas have now been treated. And buried in the molecular design of these living drugs is a detail that ties the whole field back to immune aging: nearly every clinical CAR includes a costimulatory domain, and the two dominant choices are CD28 and 4-1BB. Gilead/Kite's Yescarta uses a CD28 domain; Novartis's Kymriah — developed with Carl June's team at the University of Pennsylvania — uses 4-1BB. The very molecule that age erases is the molecule cell engineers hard-wire back in to make a T cell potent and durable.
That symmetry is not poetic license; it is a mechanism. CD28 costimulation drives the metabolic fitness and persistence that exhausted or senescent T cells lack. It is why Crystal Mackall's group at Stanford, working to keep engineered T cells from burning out inside solid tumors, showed that overexpressing the transcription factor c-Jun could render CAR T cells resistant to exhaustion — a direct attack on the dysfunction that also defines aged immunity.[21]
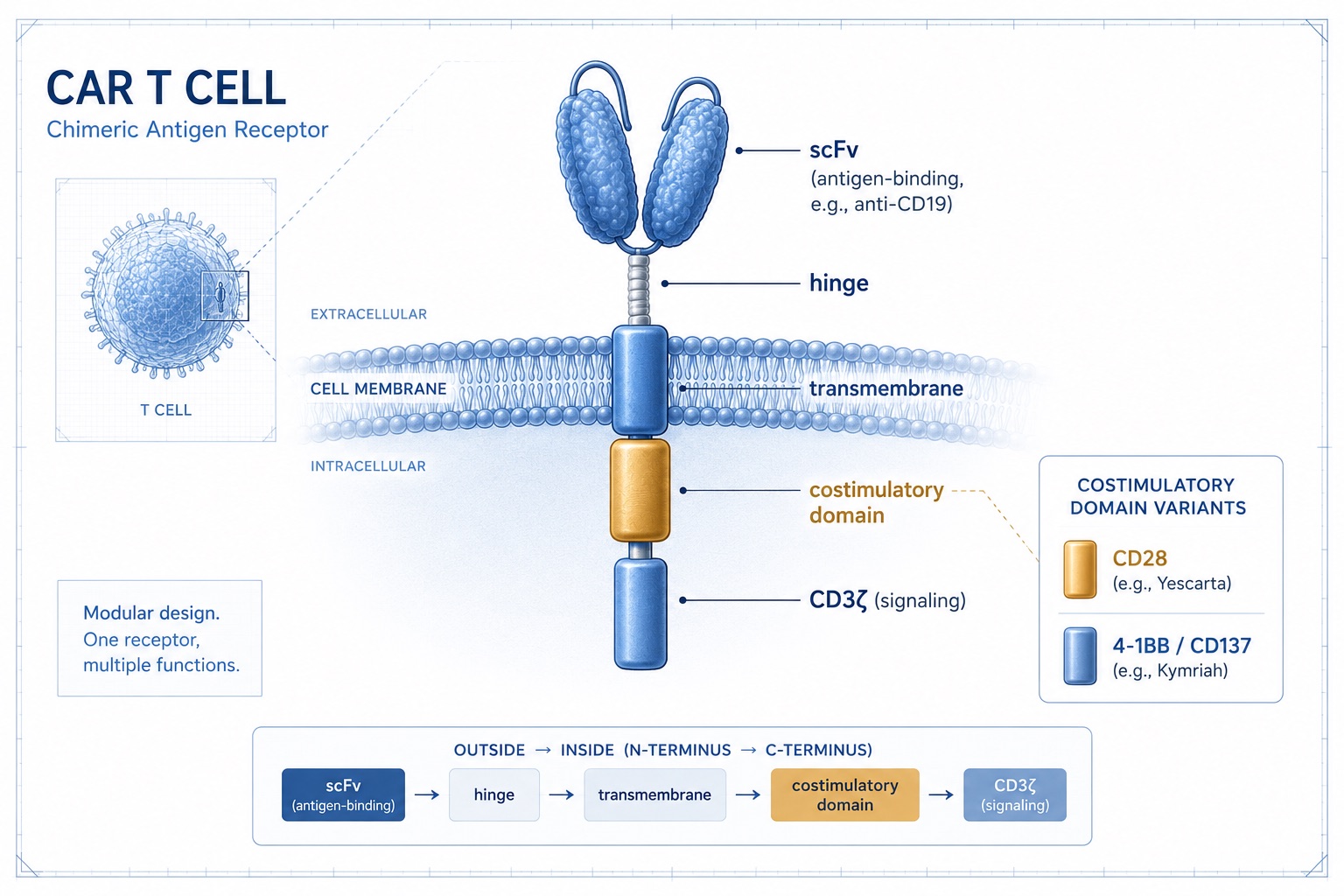
Figure 6. Chimeric Antigen Receptor (CAR) T-Cell Design — CD28 vs 4-1BB Costimulatory Domains
Cell engineers build "living drugs" by hard-wiring the very signal that age erases. Most clinical CAR T cells carry a CD28 or 4-1BB costimulatory domain — the same CD28 biology, reinstalled by design.
And then the fields fully collide. In 2024, Corina Amor, Scott Lowe, and Michel Sadelain reported in Nature Aging that senolytic CAR T cells — engineered to recognize and destroy senescent cells — produced prophylactic, long-lasting protection against age-related metabolic dysfunction in mice, building on their 2020 Nature work clearing senescent cells from fibrotic tissue.[18],[19] Read that again: a cancer technology, re-pointed at the cells of aging itself. The same engineering that gives a depleted immune system new killers can, in principle, be aimed at the senescent debris that drives inflammaging. This is the clearest existing proof of concept that the aged immune compartment is not a fixed inheritance but an engineerable system. We are, for now, decisively in the mouse — but the conceptual wall between "cancer immunotherapy" and "longevity medicine" has already fallen.
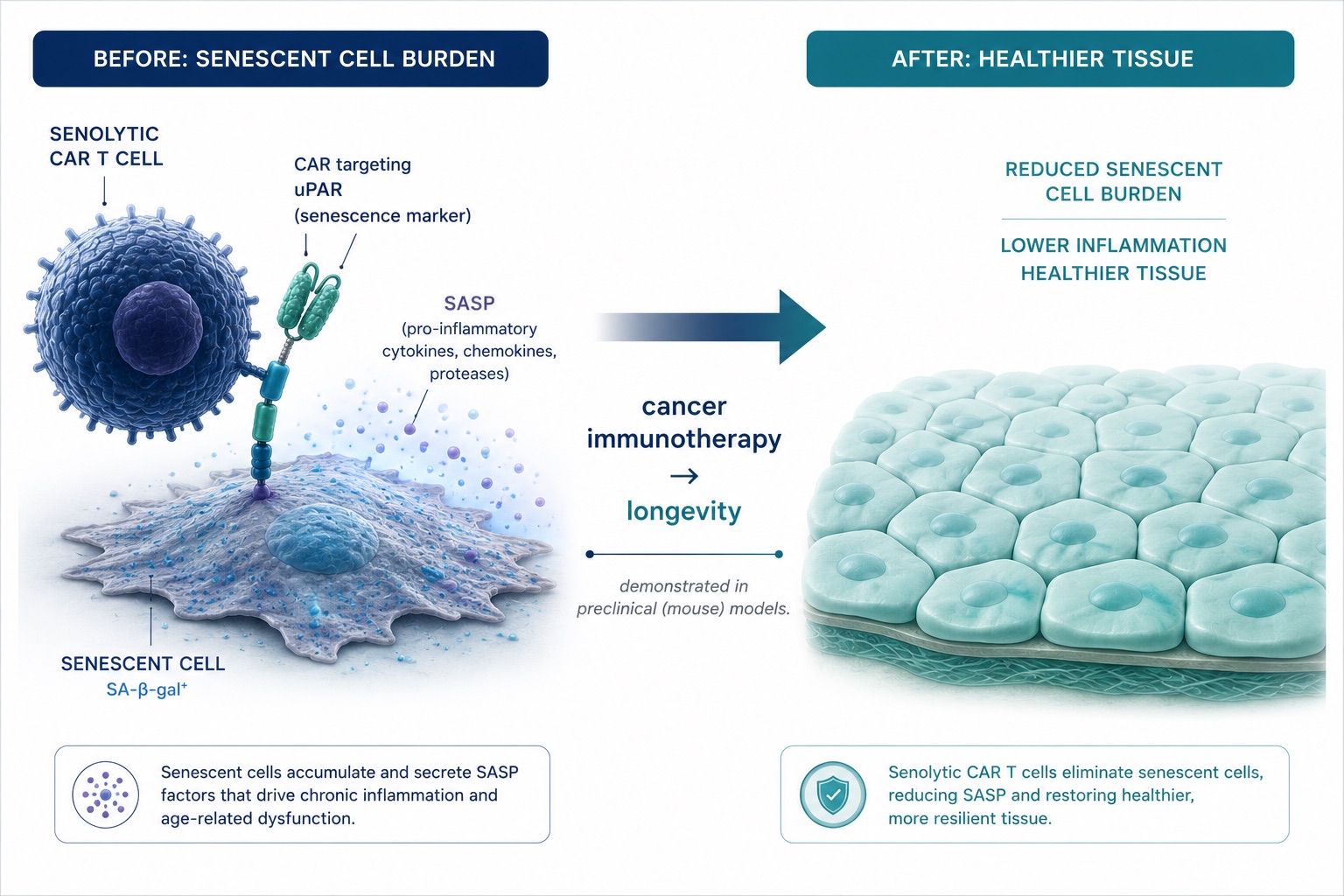
Figure 7. Senolytic CAR T Cells — Engineering Immune Cells to Clear Senescent Cells in Aging Tissue
The same technology that fights cancer is being aimed at aging itself — senolytic CAR T cells engineered to hunt and clear senescent cells. Proven in mice, it is the clearest proof-of-concept that the aged immune compartment is an engineerable system.
6. The Skeptic's Caveat: This Is a Sharp Instrument
If you are the kind of reader this article is written for, you have already spotted the catch. A signal powerful enough to rejuvenate is powerful enough to harm, and the history here is sobering.
In 2006, a CD28 superagonist antibody called TGN1412 entered a first-in-human trial and triggered a near-fatal cytokine storm in six healthy volunteers within hours. The molecule was not stupid; it was simply a reminder that CD28 sits at the center of an exquisitely balanced system. Notably, targeted CD28 agonism has since re-emerged as a more carefully engineered strategy in cancer immunotherapy, with a much deeper respect for dose and context.[17] The lesson is not "CD28 is dangerous." It is "CD28 is consequential, and consequential systems are not playgrounds for guesswork."
Two more nuances keep the picture honest. First, CD28-null cells are not uniformly villains — some CD8+CD28− populations behave as regulatory cells, and in CD4+ cells the loss is linked to the very autoimmune and cytotoxic functions that drive rheumatoid arthritis and vascular inflammation. Indeed, the drug abatacept works by blocking CD28 costimulation, and a patient's CD28-null burden can predict their response to it. More CD28 is not globally "better." Context is everything. Second, CD28 shares its B7 ligands with CTLA-4, the inhibitory receptor whose discovery by James Allison at MD Anderson — alongside Tasuku Honjo's PD-1 and Gordon Freeman's PD-L1 work — founded the entire checkpoint-blockade revolution.[22] The on-switch and the off-switch compete for the same dock. You cannot intelligently push one without understanding the other.
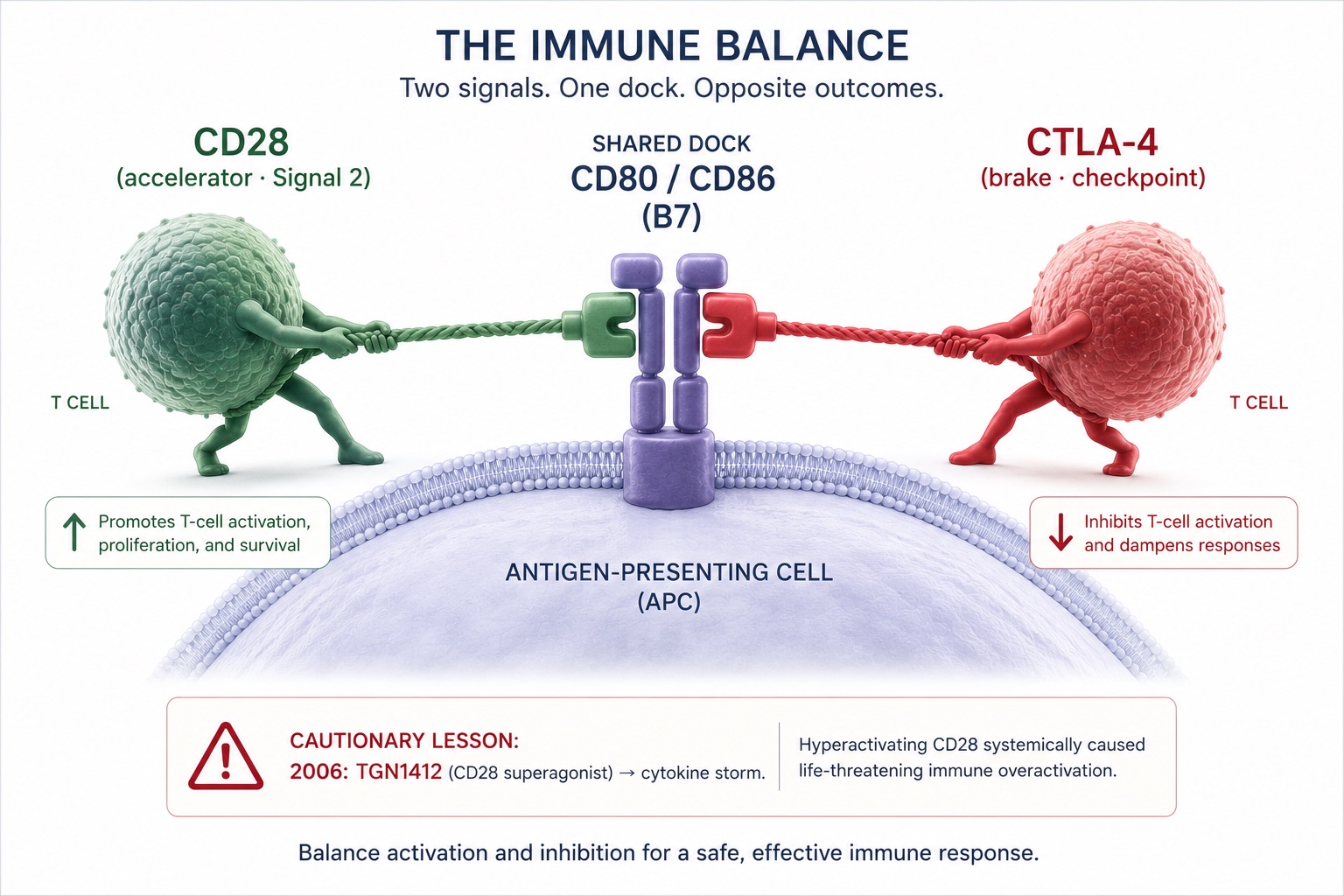
Figure 8. The CD28–CTLA-4 Balance — Competing Costimulatory and Checkpoint Signals for the Same B7 Ligands
The immune system's accelerator (CD28) and its brake (CTLA-4) compete for the same molecular dock. That shared wiring is why checkpoint drugs work — and why deliberately pushing CD28 is a job for clinicians, not guesswork.
This is precisely why immune longevity is the opposite of a do-it-yourself project, and why the responsible posture is measurement first, intervention second, and humility throughout.
7. From Frontier Science to Your Longevity Plan
So what does a rigorous person actually do with all this in 2026? Do not order a CD28 infusion — no such validated longevity therapy exists, and anyone selling one should be treated with suspicion. The honest, evidence-graded answer separates three tiers.
Tier one — measure. The most actionable insight from the CD28 literature is that immune age is quantifiable and only loosely correlated with the number on your driver's license. Immune-phenotyping panels (naïve-to-memory ratios, CD28-null fractions, CMV serostatus) are increasingly available through research-grade diagnostics, and a growing ecosystem of consumer testing platforms is racing to bring this kind of depth to the individual. Function Health and Superpower have built memberships around comprehensive biomarker panels; Lifeforce pairs longitudinal testing with clinician oversight and treatment; newer entrants like Protocole and Extension Health are extending the model. They differ in philosophy — pure diagnostics versus diagnostics-plus-protocol — but they share one premise this science endorses: a single annual snapshot is a photograph, and aging is a motion picture.
Tier two — the unglamorous interventions that actually move immune aging. Here, the data are strongest and least exciting. Chronic infection load matters: CMV status is one of the heaviest known accelerants of CD28-null accumulation. Vaccination timing matters more if you act while your repertoire is still broad. And — to the eternal irritation of those seeking a molecule — resistance training and cardiorespiratory fitness remain among the best-evidenced modifiers of immune and inflammatory aging. Muscle is an endocrine organ; movement is an anti-inflammatory drug you already own. Sleep, the substrate on which T-cell memory is consolidated, is not optional. None of this is sexy. All of it is real.
Tier three — the frontier you watch, not yet the frontier you buy. Senolytics, engineered cell therapies, thymic regeneration, and CD28-modulation belong here. They are genuinely promising and genuinely unproven for healthy-aging use in humans. A serious longevity plan holds a watching brief on tier three while executing relentlessly on tiers one and two.
The connective tissue across all three tiers is artificial intelligence — and not as a buzzword. The reason is structural: immune aging is high-dimensional. CD28 status, CMV load, inflammatory markers, sleep architecture, training load, and genetics interact in ways no clinician can hold in their head. This is exactly the problem a digital twin is built to solve. By fusing multi-modal health data — wearables, labs, medical records, and lifestyle inputs — into a personalized physiological model, a longevity platform can move from population averages to your trajectory: what your inflammatory baseline does if you add two strength sessions a week, or whether your vaccine response window is closing faster than your peers'. At LongevityPlan.AI, we describe this as a sensor layer (the data) feeding an intelligence layer (the model that turns data into a decision). The same logic now reshaping clinical trials — companies like Unlearn.AI building digital control arms, and AI-discovery platforms such as Insilico Medicine and XtalPi compressing the path from target to molecule — is the logic that turns a stack of immune biomarkers into a plan.
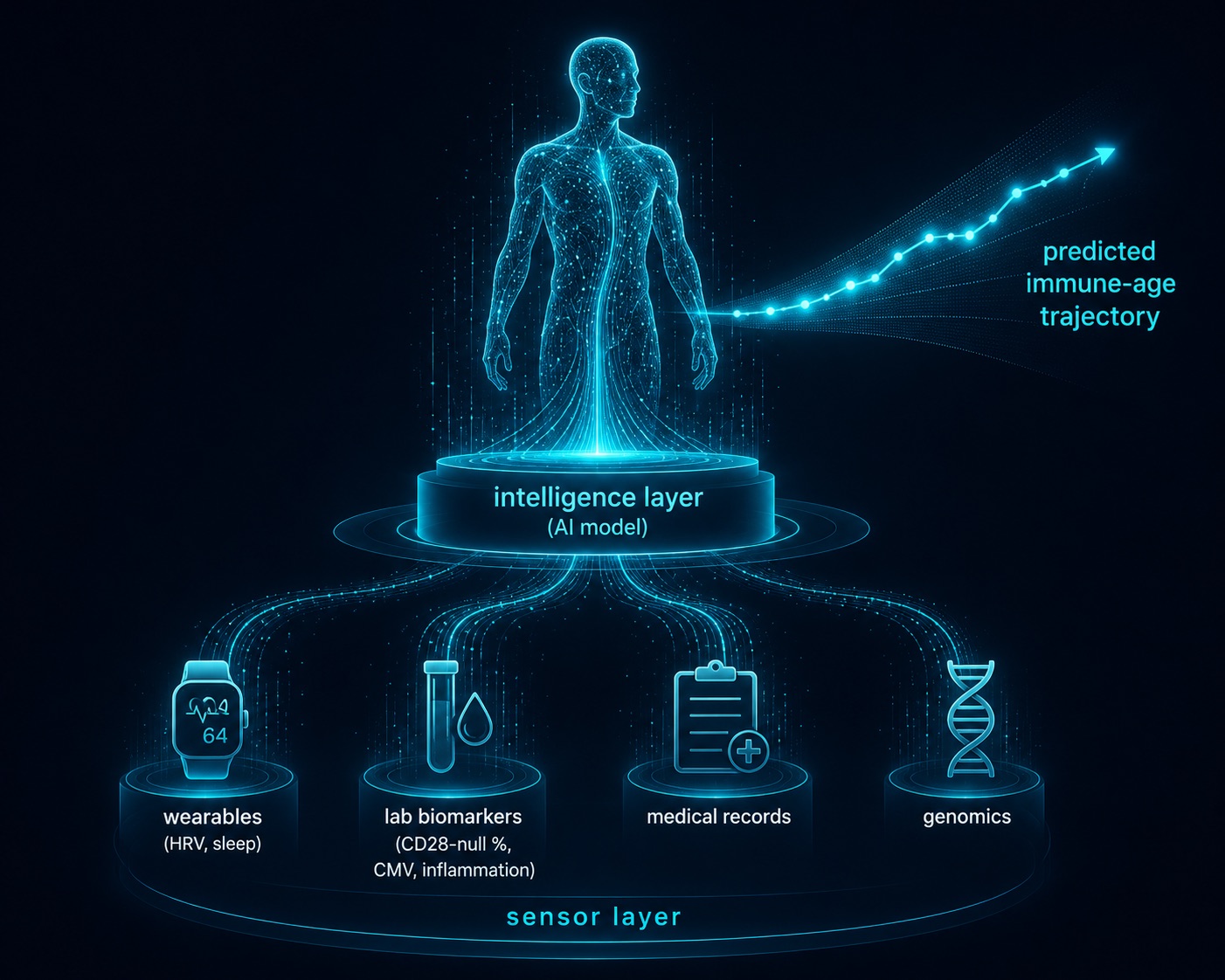
Figure 9. AI-Powered Digital Twin for Longevity — Fusing Multi-Modal Health Data Into a Predictive Immune-Age Model
Immune aging is high-dimensional — too complex for intuition. A digital twin fuses wearable, lab, and genomic data (the sensor layer) into an AI model (the intelligence layer) to forecast your trajectory, not the population average.
8. Why Your Response Won't Match Your Neighbor's
There is a final reason the averages fail you, and it is written into your DNA. Two executives can follow the identical protocol and diverge wildly, because the genes governing inflammation, detoxification, hormone metabolism, and drug response differ between them. This is the domain of pharmacogenomics, and it is the part of "personalized medicine" that is too often skipped.
It is also where peptide and intervention planning becomes genuinely individual. Whether a given regenerative or signaling peptide helps, does nothing, or causes side effects depends in part on an individual's genetic wiring — the enzymes that clear it, the receptors that sense it, the inflammatory tone it acts upon. The Genomics Company, a saliva-based genomic-education service for executives and high performers, frames this well; its founder, Jennifer Little-Fleck argues that your genome is "your operating manual," not your destiny. That is the right instinct. A genetic baseline never changes, which makes it the ideal foundation layer for a model that does: it is the bedrock beneath a Digital Twin for Predictive Peptide Performance™, the substrate that lets a system predict — rather than guess — how a specific person will respond before the first dose. Genomics tells you the terrain; the digital twin plans the route across it.
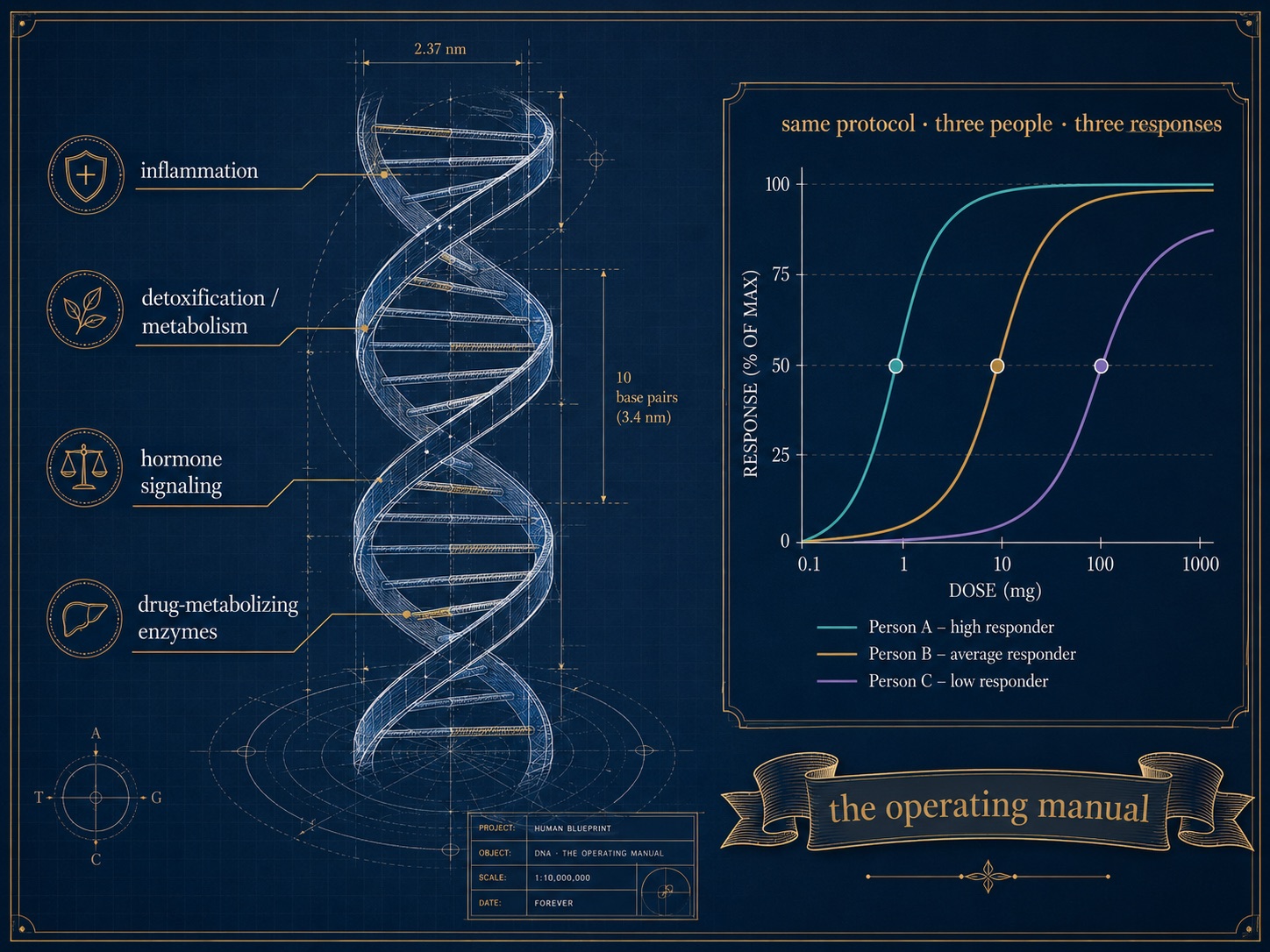
Figure 10. Pharmacogenomics and Personalized Response — Why Your Genome Predicts Individual Peptide and Drug Response
Two people on the identical protocol can diverge — because the genes governing inflammation, metabolism, and drug response differ. The genome is the fixed foundation beneath a predictive model: the operating manual, not the destiny.
This is the difference between wellness theater and precision medicine. A nutraceutical brand sells the same capsule to everyone and calls the average a result. A data-driven plan starts from the premise that you are not the average — and the CD28 story is the perfect illustration of why. Two people the same age can have immune systems a biological decade apart.
9. The Honest Bottom Line
Let's land this where the evidence allows, not where enthusiasm would prefer.
We cannot today restore CD28 to rejuvenate a human immune system on demand. There is no approved therapy, and the most exciting reversal data live in cell cultures and mice. Anyone who tells you otherwise is ahead of the science.
But the stronger, defensible claim survives intact: the aging of the immune system is no longer a black box, and the loss of CD28 is increasingly understood as a regulated, measurable, and — at least in the laboratory — modifiable process rather than an immutable verdict. Engineers already rebuild CD28 signaling routinely to create the most powerful cancer drugs we have. Those same tools are being aimed, in the lab, at aging itself. And the levers that demonstrably affect immune aging in humans today — fitness, sleep, infection management, vaccination strategy, and, above all, measurement — are available to anyone willing to trade guesswork for data.
That is the philosophy behind precision longevity, and it is fundamentally optimistic. You are not at the mercy of a clock you cannot see. The clock is readable. The question worth asking your Coach / Practitioner at your next review is not "how do I feel?" but "what is my immune age, and what is the plan to defend it?" The first generation to ask that question systematically — to bring an athlete's discipline and an engineer's data to their own biology — may well be the first to bend the curve. Science says the dial exists. The rest is execution.
Sign Up for FREE Daily Longevity Club Workshops
Sign Up for the LongevityPlan Affiliate Program (Pays 20%)
About the Author
Tony Medrano is CEO and Co-Founder of LongevityPlan.AI, an AI-driven platform that unifies wearable, laboratory, clinical, and lifestyle data into a personalized digital twin for healthspan optimization. A three-time Ironman triathlete and former U.S. Navy officer with degrees from Harvard, Columbia, and a JD/MBA from Stanford, he writes on AI-powered preventive medicine and the science of human performance.
This article is educational and is not medical advice; immune and cell-based interventions discussed here are investigational and should only be considered with qualified clinicians.
Endnotes
- Esensten JH, Helou YA, Chopra G, Weiss A, Bluestone JA. CD28 Costimulation: From Mechanism to Therapy. Immunity. 2016;44(5):973–988. PMID: 27192564.
- Weng NP, Akbar AN, Goronzy J. CD28− T cells: their role in the age-associated decline of immune function. Trends Immunol. 2009;30(7):306–312. PMID: 19540809.
- Vallejo AN. CD28 extinction in human T cells: altered functions and the program of T-cell senescence. Immunol Rev. 2005;205:158–169. PMID: 15882352.
- Vallejo AN, Bryl E, Klarskov K, Naylor S, Weyand CM, Goronzy JJ. Molecular basis for the loss of CD28 expression in senescent T cells. J Biol Chem. 2002;277(49):46940–46949. PMID: 12324461.
- Warrington KJ, Vallejo AN, Weyand CM, Goronzy JJ. CD28 loss in senescent CD4+ T cells: reversal by interleukin-12 stimulation. Blood. 2003;101(9):3543–3549. PMID: 12506015.
- Effros RB, et al. Loss of CD28 expression on T lymphocytes: a marker of replicative senescence. Dev Comp Immunol. 1998;22(5–6):523–532. PMID: 9463780.
- Effros RB. The role of CD8+ T-cell replicative senescence in human aging. Immunol Rev. 2005;205:147–157. PMID: 15882351.
- Goronzy JJ, Weyand CM. Mechanisms underlying T cell ageing. Nat Rev Immunol. 2019;19(9):573–583.
- Mittelbrunn M, Kroemer G. Hallmarks of T cell aging. Nat Immunol. 2021;22(6):687–698. PMID: 33986548.
- Desdín-Micó G, et al. T cells with dysfunctional mitochondria induce multimorbidity and premature senescence. Science. 2020;368(6497):1371–1376.
- Akbar AN, Henson SM. Are senescence and exhaustion intertwined or unrelated processes that compromise immunity? Nat Rev Immunol. 2011;11(4):289–295. PMID: 21436838.
- Akbar AN, Henson SM, Lanna A. Senescence of T lymphocytes: implications for enhancing human immunity. Trends Immunol. 2016;37(12):866–876. PMID: 27720177.
- Pereira BI, De Maeyer RPH, Covre LP, et al. Sestrins induce natural killer function in senescent-like CD8+ T cells. Nat Immunol. 2020;21(6):684–694.
- Lewis DE, Merched-Sauvage M, Goronzy JJ, Weyand CM, Vallejo AN. Tumor necrosis factor-α and CD80 modulate CD28 expression through a similar mechanism of T-cell receptor-independent inhibition of transcription. J Biol Chem. 2004;279(28):29130–29138. PMID: 15128741.
- Brunner S, et al.; and Vallejo AN, et al. — Epigenetic/microRNA regulation of CD28: see Involvement of MicroRNAs in the Aging-Related Decline of CD28 Expression by Human T Cells. Front Immunol. 2018;9:1400 (PMC6015875); and whole-genome DNA-methylation profiling of CD28-null T cells, Aging Cell (PMC5334526).
- Lin Y, et al. CD28null T cells in aging and diseases: From biology to assessment and intervention. Int Immunopharmacol. 2024;131:111807. PMID: 38471362.
- Edner NM, et al. CD28 co-stimulation: novel insights and applications in cancer immunotherapy. Nat Rev Immunol. 2024 (online) — reviews the 2006 TGN1412 superagonist trial and the re-emergence of targeted CD28 agonism.
- Amor C, et al. Senolytic CAR T cells reverse senescence-associated pathologies. Nature. 2020;583(7814):127–132.
- Amor C, Fernández-Maestre I, ..., Sadelain M, Lowe SW. Prophylactic and long-lasting efficacy of senolytic CAR T cells against age-related metabolic dysfunction. Nat Aging. 2024;4(3):336–349.
- June CH, Sadelain M. Chimeric Antigen Receptor Therapy. N Engl J Med. 2018;379(1):64–73.
- Lynn RC, Weber EW, ..., Mackall CL. c-Jun overexpression in CAR T cells induces exhaustion resistance. Nature. 2019;576(7786):293–300.
- Leach DR, Krummel MF, Allison JP. Enhancement of antitumor immunity by CTLA-4 blockade. Science. 1996;271(5256):1734–1736.
- Strioga M, Pasukoniene V, Characiejus D. CD8+CD28− and CD8+CD57+ T cells and their role in health and disease. Immunology. 2011;134(1):17–32.
- Frasca D, et al.; and reviews on immunosenescence in rheumatoid arthritis (PMC5388800) describing CD28-null burden as a predictor of response to CD28-blocking therapy (abatacept).